T-cell immunotherapy, particularly chimeric antigen receptor (CAR) T-cell therapy, has emerged as a revolutionary approach in cancer treatment. The success of CAR T-cells in treating B-cell malignancies has spurred extensive research into expanding its application to other cancers and enhancing both its efficacy and safety. The modular design of CARs, comprising an extracellular antigen-binding domain, a transmembrane domain, and intracellular T-cell signaling domains, allows for iterative optimization and the creation of Next-gen Vehicle Models in therapeutic CAR design. This review delves into the significant recent progress in CAR engineering and explores how these advancements are shaping the future of clinical applications for this groundbreaking therapy.
Immunotherapies that utilize genetically modified T cells, most notably those expressing chimeric antigen receptors (CARs) to redirect T-cell specificity, represent a significant leap forward in cancer treatment. These personalized, living therapeutics, while holding immense promise, present unique challenges in development and optimization. This article provides an overview of the critical aspects of CAR T-cell design, highlighting key findings that have propelled the field to its current state and are likely to define its future trajectory in the realm of next-gen vehicle models for targeted therapy. This is not intended to be an exhaustive compilation but rather a focused perspective on the most impactful discoveries shaping the evolution of CAR T-cell technology.
T-cell receptor (TCR) specificity dictates a T cell’s target recognition capabilities.1 T cells are crucial in immune surveillance, identifying and eliminating infected cells through TCR-mediated detection of microbial antigens presented by major histocompatibility complex (MHC) proteins. TCR engagement with a specific MHC-peptide complex initiates a cascade of intracellular signals, starting with the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) domains within TCR accessory proteins such as CD3ζ, CD3γ, CD3δ, and CD3ε.2,3 This signaling cascade culminates in T-cell activation and the destruction of the target cell. T cells can also target cancerous cells by recognizing tumor antigens, which may be novel, germ cell-specific, or mutated self-antigens (neo-epitopes).4 The efficacy of tumor-reactive T cells has been validated through the isolation of tumor-infiltrating lymphocytes (TILs) from metastatic cancer patients, their ex vivo expansion, and subsequent reinfusion into patients.4,5 This approach has led to durable complete remissions (CRs) in some patients, outcomes rarely seen with conventional cytotoxic chemotherapies.6 However, the widespread adoption of TIL therapy is hampered by the lengthy and labor-intensive process (2–3 months) required to generate a sufficient number of tumor-reactive T cells.7 CAR technology provided a critical breakthrough, enabling the rapid generation of large populations of tumor-reactive T cells within approximately one week and achieving positive clinical outcomes in numerous patients with acute or chronic leukemia.8,9 This speed and efficiency are key advantages in the development of next-gen vehicle models for rapid therapeutic response.
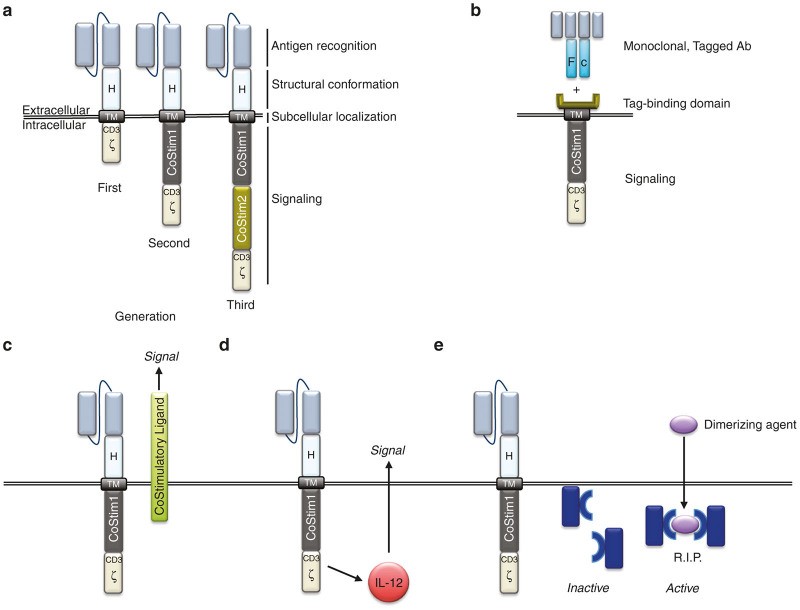
The CAR is a synthetic antigen receptor, a fusion of antibody and TCR components, consisting of an extracellular antigen-binding domain and intracellular signaling domain(s) (Figure 1a).10 Genetically modifying a T cell with a CAR imparts a new antigen specificity via the single-chain variable fragment (scFv), derived from a tumor-specific antibody.1 The scFv allows the T cell to bind to a tumor antigen, triggering T-cell activation through intracellular domains originating from CD3ζ ITAM domains.1,3 A hinge and a transmembrane domain (TM), typically from CD8α or immunoglobulin, connect the extracellular scFv and intracellular CD3ζ ITAM domains, completing the CAR genetic construct. Early in vitro studies demonstrated that T cells engineered with first-generation CARs, possessing only a CD3ζ intracellular signaling domain, could activate and kill target cells. However, these first-generation CAR T cells exhibited limited persistence and antitumor efficacy in vivo.11 The modularity of CAR technology allows for continuous refinement to optimize T-cell function, leading to the evolution from first-generation to second-generation CARs, marking a significant step in the development of next-gen vehicle models.
Figure 1.
 Figure 1
Figure 1Schematic representation of Chimeric Antigen Receptor (CAR) designs and accessory strategies for enhanced CAR T-cell therapy. (a) Basic CAR structure illustrating the antigen recognition domain (scFv), hinge (H) or spacer region, transmembrane domain (TM), and intracellular signaling domains (CD3ζ for first-generation, plus costimulatory domain for second-generation, and two costimulatory domains for third-generation CARs). (b) Universal CAR design where the signaling domain binds to a tag (e.g., fluorescein isothiocyanate or biotin) on a monoclonal antibody (Ab), allowing for antigen specificity to be determined by the antibody used. (c) Trans signaling approach where costimulatory ligands are co-expressed to stimulate neighboring cells within the immune synapse. (d) Cytokine gene co-expression under inducible promoters (e.g., NFAT-responsive promoters for IL-12 production) to modulate the tumor microenvironment. (e) Incorporation of safety switch genes, such as inducible caspase 9 (iCasp9), to enable controlled elimination of CAR T-cells in case of severe toxicity.
TCRs are highly specific to short peptide sequences (8–12 amino acids) from foreign antigens, which carries a risk of cross-reactivity with similar host antigen sequences.12 TCR ligation of self-antigens can trigger T-cell activation, potentially leading to autoimmunity and even fatal outcomes. To mitigate this risk, T cells typically require two signals for full activation.13 The primary signal is delivered through the TCR, while the secondary, costimulatory signal is mediated by CD28 ligation with CD80 or CD86, molecules normally expressed on antigen-presenting cells (APCs).12,13 Thus, if a T cell encounters a cross-reactive peptide on a normal (non-APC) cell, the absence of costimulation prevents full T-cell activation. However, activated APCs, such as during inflammation or infection, upregulate CD80 and CD86, providing both signal 1 and signal 2, thereby enabling complete T-cell activation, target cell killing, and long-term persistence.12,13 CAR researchers adopted this two-signal model by engineering second-generation CARs to include a CD28 costimulatory domain in tandem with CD3ζ ITAM domains (Figure 1a).14–16 These second-generation CARs not only enhanced in vitro T-cell activation and killing but also significantly improved in vivo tumor eradication and T-cell persistence. Furthermore, costimulatory domains beyond CD28, including CD27, 4-1BB, and OX40, have been shown to provide similar enhancements to CAR T-cell function and persistence in vivo.17–19 These advancements in costimulatory domains are crucial for developing effective next-gen vehicle models.
Second-generation CAR T cells have demonstrated remarkable antileukemia activity in phase 1 clinical trials. Complete remission rates as high as 90% have been achieved in patients with relapsed and/or refractory B-cell acute lymphoblastic leukemia (B-ALL) treated with second-generation CD19-targeted T cells incorporating a CAR with a 4-1BB or CD28 costimulatory domain.20–22 While the success of CD19 targeting is undeniable, concerns regarding safety and efficacy arise when applying this technology to other cancers. Recognizing these challenges, researchers have continued to leverage the modular nature of CARs to further refine and optimize this novel antigen-receptor. The following sections will explore how modifications to the scFv, hinge/spacer, and intracellular domains are being engineered to create safer and more effective next-gen vehicle models for cell-based cancer therapy.
The Single Chain Variable Fragment
The scFv component of the CAR is responsible for redirecting a bulk population of autologous T cells to recognize a new tumor antigen. However, recent studies have revealed that the scFv’s influence extends beyond target specificity, significantly impacting CAR function, safety, and efficacy. For instance, the nonhuman origin of antibodies used to create scFvs can trigger anti-CAR immune responses, potentially limiting the persistence of infused CAR T cells.23 More seriously, such immune responses have been implicated in fatal anaphylaxis in patients receiving multiple doses of CAR T cells.24 Consequently, while current leading CARs for B-cell malignancies utilize murine-derived scFvs, future CARs targeting other cancers in development are increasingly being “humanized” to minimize these immunogenic risks. This shift towards humanized scFvs is a key aspect of developing safer next-gen vehicle models.
The affinity of the scFv for its target antigen is another critical parameter being optimized to enhance gene-targeted T-cell function. CD19 has proven to be an ideal target for demonstrating the proof-of-principle of CAR T-cell therapy. However, the experience with CD19 targeting has also highlighted the concern of on-target/off-tumor toxicity, as patients have experienced prolonged B-cell aplasia.21,25,26 While B-cell aplasia is generally manageable with gamma globulin and/or antibiotics, targeting antigens expressed on vital tissues, such as ERBB2 on respiratory epithelium, has resulted in fatal toxicities.27 As CARs are being developed for solid tumors, where target antigens are often shared with normal epithelial tissues, the risk of severe complications increases. However, recent research has shown that differential antigen expression levels and scFv affinity can be exploited to discriminate between tumor cells and normal tissues. Low-affinity scFvs have been demonstrated to effectively mediate CAR T-cell killing of tumor cells with high antigen expression while sparing normal cells with low or normal antigen levels.28,29 This antigen-density-dependent discrimination, observed both in vitro and in vivo, suggests that optimizing scFv affinity can significantly improve the safety profile of CAR T-cells, particularly when targeting antigens present on healthy tissues. This affinity modulation is a crucial strategy for creating safer next-gen vehicle models.
A significant advancement in CAR design was the realization that incorporating both CD3ζ (signal 1) and CD28 costimulation (signal 2) into a single CAR construct could effectively mimic normal TCR activation and costimulation.14–16 Further innovation has explored the use of bispecific CARs, requiring combinatorial ligation of two distinct tumor antigens to enhance safety and maintain potent cancer-killing activity.30–32 This approach involves dissociating the activation and costimulatory domains onto separate, complementary CARs. CAR1, for example, might contain only the CD3ζ activation domain, while CAR2 contains only costimulatory domains. Ligation of either CAR1 or CAR2 alone is insufficient for full T-cell activation, delivering only one signal. However, when both CAR1 and CAR2 simultaneously engage their respective antigens, CD3ζ (signal 1) and costimulation (signal 2) are delivered in concert, enabling T-cell activation and sustained in vivo function. Beyond this design, other dual CAR systems have been developed to enhance tumor targeting safety and/or efficacy.30–32 For instance, a secondary CAR specific for a normal tissue antigen can be engineered to express a suppression domain, preventing the destruction of healthy tissue.33 This inhibitory bispecific CAR strategy is being explored to prevent off-tumor toxicities.33 The demonstration that combinatorial antigen ligation and signal domain dissociation can regulate T-cell activation and function provides a robust platform for future CAR design manipulations, paving the way for next-gen vehicle models with enhanced safety and precision.
Beyond scFv optimization for safety, researchers have explored replacing the scFv entirely to create ligand-based or universal CARs. Ligand-based CARs substitute the scFv with a ligand for a tumor marker; for example, a CAR expressing a ligand for the IL13 receptor (IL13R) can redirect T cells to IL13R-expressing glioblastoma cells.35 While ligands may sometimes exhibit multiple binding partners, ligands used in CAR design, like the IL13-zetakine, are often engineered for high specificity to the target receptor, such as IL13R in glioblastoma. Universal CAR systems represent another innovative approach to broaden the clinical utility of CAR T-cell therapy (Figure 1b). These universal CARs can retarget patient T cells to mediate antitumor responses irrespective of cancer type. This technology involves replacing the scFv with a binding domain specific for a tagged molecule, such as biotin or fluorescein isothiocyanate.36,37 The extracellular portion of the universal CAR, linked to a transmembrane domain and intracellular signaling domains (costimulatory and activation domains), can then bind to a tumor-specific antibody labeled with the tag.Figure 1b When a tumor-specific, tagged antibody binds its target on a tumor cell, the universal CAR can then engage the tagged antibody, triggering T-cell activation and tumor cell killing.36,37 Both universal CAR T-cell systems have shown functional validation in animal models.36,37 Although universal CAR T-cell technology is still in preclinical development, it offers the significant advantage of compatibility with a wide range of clinically approved and investigational tumor-specific antibodies. Universal CARs represent a versatile platform for next-gen vehicle models adaptable to diverse cancer targets.
Hinge and Spacer Domains
The hinge, spacer, and transmembrane domains play a crucial role in linking the scFv to the intracellular activation domains and anchoring the CAR within the T-cell membrane.1 Recent studies have demonstrated that these domains are not merely structural elements but can significantly impact CAR T-cell function, offering opportunities for optimization to enhance antigen binding and T-cell signaling. Specifically, spacers have been shown to be essential for effective binding to membrane-proximal epitopes and supporting efficient target cell killing.38–40 Conversely, spacers can diminish CAR function when the epitope is located near the amino-terminal region of the cell surface protein.40 However, spacer length is not the sole determinant of function. Researchers have also found that incorporating Fc domains within the hinge or spacer region can lead to unintended antibody binding and immune cell activation.41–44 Due to the abundance of antibodies in vivo, these Fc-binding CARs can induce excessively strong and persistent T-cell activation, resulting in activation-induced cell death or the activation of other immune cells that suppress CAR T-cell persistence. Removing these Fc binding sites has been shown to enhance in vivo CAR T-cell function and persistence.41–44 These findings indicate that CAR hinge and spacer length and sequence should be carefully optimized based on the epitope’s position relative to the cell membrane. It is important to note that much of this research has focused on immunoglobulin-derived hinge and spacer regions, and further investigation is needed to fully evaluate the impact of CD8-derived spacers and hinges. Optimizing hinge and spacer domains is a critical aspect of fine-tuning next-gen vehicle models for optimal antigen engagement.
Intracellular Domains
Currently, clinical CAR T-cell therapies primarily utilize intracellular signaling domains derived from CD28, 4-1BB (CD137), or combinations thereof. However, preclinical research has explored a broader range of alternatives. A direct clinical comparison between first-generation and second-generation CARs was conducted by Savoldo and colleagues. In this study, patients received infusions of both first-generation CAR T cells and second-generation CAR T cells (containing a CD28 costimulatory domain), both targeting the same antigen. Post-infusion persistence analysis using polymerase chain reaction revealed that CD28-containing CAR T cells significantly outperformed their first-generation counterparts. This study provided definitive clinical evidence that CD28 costimulation enhances CAR T-cell survival in patients.45 This clinical validation underscored the importance of costimulatory domains in next-gen vehicle models.
While CD28 costimulation is crucial for the clonal expansion of activated T cells and IL-2 secretion in the early phases of activation, another costimulatory receptor, 4-1BB (CD137), is associated with promoting long-term T-cell survival. Costimulatory domains from 4-1BB have been incorporated into both second-46 and third-[47](#bib47] generation CARs. The inclusion of 4-1BB costimulation has been shown to mitigate exhaustion caused by tonic CD3-signaling observed in CD28 CARs.48 Beyond CD28 and 4-1BB, OX40 (CD134), a CD4-associated costimulatory receptor, has also been preclinically evaluated as a CAR signaling component. A third-generation CAR incorporating both CD28 and OX40 costimulatory domains demonstrated superior survival of CCR7(-) T cells compared to a CD28-only CAR.49 Furthermore, a CD28-OX40 CAR induced less IL-10 secretion than a CD28-based second-generation CAR, without affecting IFN-γ secretion.50
ICOS, a costimulatory receptor involved in TH17 polarization, has also been integrated into the intracellular domain of CARs. Its incorporation was associated with enhanced in vivo persistence of TH17-polarized CAR T cells compared to CD28 or 4-1BB costimulation.51 Similarly, CD27 costimulation has been shown to provide better survival than CD28.18 In an elegant study by Duong and colleagues, a combinatorial approach involving random generation and selection of intracellular signaling domains was employed. Screening a library of CAR constructs with varying numbers of costimulatory domains, they identified a third-generation design containing DAP10 and CD27, in addition to CD3ζ, which exhibited the greatest antitumor effect.52
More recently, a CAR incorporating a DAP12-derived signaling domain was developed for NK cell-based adoptive therapy, demonstrating more robust performance than CD3ζ-based CARs.53 In T cells, a “KIR-CAR,” consisting of an scFv fused to the transmembrane domain of KIR2DS2, along with DAP12 as a separate molecule, showed superior antitumor efficacy in vivo compared to a second-generation, CD3ζ-based CAR. This novel design also exhibited enhanced surface expression compared to its CD3ζ counterpart, potentially due to differences in membrane dynamics, which could impact CAR T-cell function.54 Exploring diverse intracellular signaling domains is key to optimizing the function of next-gen vehicle models.
A comprehensive understanding of the molecular pathways triggered by CARs, both with and without antigen ligation, is essential for rationally designing the most effective receptors. This will require the application of systems biology approaches in preclinical experimentation and the integration of molecular, biological, and pharmacodynamic data from clinical analyses of gene-modified T cells. This holistic approach is crucial for the development of truly advanced next-gen vehicle models.
Joint Expression of CAR and Accessory Genes
CARs have also been combined with accessory proteins to enhance T-cell function and/or utilize lymphocytes as delivery vehicles for payloads that modify the tumor microenvironment. T cells engineered to express a tumor-specific CAR alongside a chemokine receptor that binds chemokines present in the tumor microenvironment exhibit improved tumor targeting and modulation of CAR T-cell homing and trafficking.56 This dual-protein system enables both dual targeting and functional modification. In another approach using accessory proteins, Zhao et al. demonstrated that a second-generation CAR with a CD28 costimulatory domain and CD3ζ, co-expressed with 4-1BBLigand in trans (CD28ζ/41BBL), induced a more potent antitumor effect than a third-generation CAR incorporating both CD28 and 4-1BB in cis (Figure 1c). Furthermore, T cells expressing a CAR with the CD28ζ/41BBL domains showed reduced exhaustion markers and prolonged in vivo persistence in a mouse model of B-cell leukemia.57 Conversely, Condomines et al. reported that the second-generation 28Z design was more effective than a first-generation CAR supplemented in trans by CD80 costimulation. This observation was associated with decreased sensitivity to CTLA4-mediated inhibition in T cells expressing the second-generation CAR,58 suggesting that different costimulatory ligands may lead to varying outcomes. Finally, CAR T-cells co-expressing CD40L also outperformed second-generation CAR T-cells. This particular design, applied to B-cell malignancies, enhanced CAR T-cell function through multiple mechanisms, including stimulation of IL-12 secretion by dendritic cells and induction of costimulatory molecules by CD40-expressing target cells.59
Cytokines have also been investigated as combination partners to potentially enhance CAR T-cell activity and/or persistence, leading to improved antitumor effects. Hoyos et al. explored the combination of a second-generation CAR targeting CD19 with interleukin-15 (IL-15) and an inducible suicide gene (iCasp9). This CAR/cytokine/suicide gene trifecta enhanced proliferation, reduced PD-1 expression, and improved anticancer killing in a humanized mouse model of B-cell lymphoma.60 In another study, Markley and Sadelain showed that anti-CD19 CAR T-cells constitutively expressing IL-2, IL-7, IL-15, or IL-21 exhibited greater antilymphoma activity in vivo compared to CAR T-cells without cytokine expression, with IL-7 and IL-21 demonstrating the most significant efficacy.61
Interleukin-12 (IL-12) is a pleiotropic cytokine capable of inducing potent antitumor responses by stimulating the immune system at multiple levels. However, systemic administration of IL-12 causes severe toxicity, limiting its clinical application.62 To overcome this limitation, systemically administered CAR T-cells can be employed as “Trojan horses” to deliver IL-12 directly to the tumor site (Figure 1d). Using this approach, Pegram and collaborators63 demonstrated that IL-12 expression by CAR T-cells eliminated the need for lymphodepleting preconditioning and rendered CAR T-cells resistant to regulatory T-cell inhibition in a syngeneic mouse model of CD19+ malignancies. Furthermore, their results indicated that IL-12’s mechanism of action was dependent on both CD8+ and CD4+ T-cell subsets and functional IFN-γ secretion.63 IL-12 secretion can be further localized to sites of T-cell activation by linking its expression to signaling pathways activated upon CAR engagement. For example, Zhang and colleagues developed γ-retroviral vectors where IL-12 expression in transduced T cells is controlled by a nuclear factor of activated T cells (NFAT)-responsive promoter.64 Utilizing this transcriptional control strategy, Chmielewski et al. demonstrated that IL-12 secretion by anti-carcinoembryonic antigen CAR T-cells resulted in the elimination of both carcinoembryonic antigen-expressing and -negative tumor cells. This indirect effect was mediated by the activation of a potent macrophage-driven, TNF-α-dependent cytotoxic mechanism.65 Similarly, Chinnasamy et al. showed that T cells targeting the tumor vasculature, also expressing NFAT-driven IL-12, modified the tumor microenvironment by eliminating myeloid-derived suppressor cells.66 Combining CARs with cytokines represents a powerful strategy for developing next-gen vehicle models with enhanced therapeutic potency.
To date, IL-12 expression by CAR T-cells has been extensively explored in preclinical models, providing valuable insights into the biological mechanisms underlying IL-12-driven immunotherapy. However, the clinical efficacy of CAR T-cells armed with IL-12 is still under investigation. A clinical trial of adoptive transfer of TIL expressing NFAT-driven IL-12 in melanoma patients showed objective responses in patients treated with T-cell infusions containing significantly fewer cells than conventionally used in TIL trials. Moreover, treatments were administered without preparative chemotherapy, and notably, a patient who had previously failed TIL treatment experienced an objective response upon treatment with TIL+NFAT-IL-12 (ref. 67). These findings suggest that IL-12 expression by adoptively transferred T cells may offer clinical benefits, but randomized clinical trials comparing combination treatments to CAR T-cells alone are needed to provide definitive answers.
Accessory molecules can also be incorporated into CAR T-cells to serve as safety switches, enabling the elimination of CAR T-cells from circulation in the event of T-cell-mediated toxicity. This approach was initially explored in hematopoietic stem cell transplantation to mitigate graft-versus-host disease. Early designs involved expressing a protein that metabolized an inactive prodrug into a cytotoxic metabolite specifically within gene-modified cells (Figure 1e). Another strategy involves co-expressing a membrane-bound protein (typically a truncated receptor lacking the intracellular signaling domain) that can be efficiently targeted by an antibody, leading to the depletion of cells expressing this protein. For instance, Wang et al. 68 described the use of a truncated form of the human epidermal growth factor receptor (huEGFRt) in adoptively transferred cells, which can be subsequently ablated by systemic treatment with a clinical-grade antibody against epidermal growth factor receptor. A comprehensive review of various suicide genes developed to date has been recently published,69 thus we will focus on a more recent technology currently under clinical evaluation: inducible caspase-9 (iCasp9). This system involves expressing a synthetic construct composed of an incomplete proapoptotic caspase-9 (lacking its caspase recruitment domain) fused to a mutated peptide derived from the FKBP12 protein. Interaction of the mutated FKBP12 domain with a small molecule induces dimerization of the fusion protein, triggering caspase-9-induced apoptosis (Figure 1e).70 Clinical testing has demonstrated the efficacy and safety of the iCasp9 system in the context of allogeneic stem cell transplantation, where the suicide gene was expressed in donor T cells adoptively transferred to patients. Patients who developed graft-versus-host disease after transplantation were treated with intravenous administration of a clinical-grade CID (AP1903), resulting in rapid and near-complete elimination of circulating donor T cells.71 Synthetic control switches like the iCasp9 system are likely to be widely used in clinical trials of adoptive immunotherapy, both for removing toxic CAR T-cells and for more refined, dose-dependent control of CAR T-cell activity, as recently described by Wu and collaborators.72 In this elegant work, the authors generated a dissociated CAR where the antigen-binding and transmembrane domains were expressed as one protein, and the intracellular signaling domains as a separate protein. Both subunits contained a heterodimerization module that, in the presence of a small molecule, assembled the CAR into a fully functional unit. Thus, CAR T-cell activation becomes dependent on two input signals: (i) antigen binding and (ii) the presence of a dimerizing agent. This design allows for real-time and dose-dependent control of CAR T-cell activity. Incorporating safety switches and control mechanisms is paramount for the development of safe and controllable next-gen vehicle models.
Future Directions
Approximately two decades after its inception, genetically engineered synthetic immune receptors expressed in T cells have proven to be an effective treatment for cancer patients. Despite achieving this significant milestone, the field is still in its early stages, and the coming years are expected to witness a substantial increase in scientific publications reporting clinical successes with CAR T-cells. Preclinical testing has been a crucial factor in the clinical success achieved in recent years, providing valuable insights such as the necessity of integrated costimulation for optimal CAR T-cell performance. Preclinical research will continue to play a vital role in advancing this field, now complemented by data from clinical samples of CAR T-cell-treated patients. Efforts will be focused on translating the clinical success of CD19-targeted therapies to other antigens and, most importantly, to solid tumors. This endeavor will require a multidisciplinary approach involving clinicians, clinical laboratories, and basic and translational researchers. Building upon the foundations of the genomic era, CAR design is anticipated to evolve beyond simple antigen-binding-activated T-cell switches to more sophisticated biological engineering constructs. Gene-modified T cells are poised to become therapeutic agents with complex sensing and effector functions, integrating information from tumor cells and the tumor microenvironment to enhance potency and specificity. The combination of CAR T-cells with other treatment modalities, such as oncolytic virotherapy, remains largely unexplored, and preclinical modeling will be instrumental in identifying optimal combination strategies to achieve synergism among multiple modalities. At the cellular level, detailed analysis of the molecular events governing CAR T-cell function and persistence will provide the essential building blocks for developing the next generation of T-cell therapies, representing the true next-gen vehicle models for targeted cancer treatment.
Marco L Davila receives research funding from the H. Lee Moffitt Cancer Center and Research Institute, American Society of Hematology, and Damon Runyon Cancer Research Foundation. He receives compensation from Celyad and Geneius Biotechnology as a consultant. He receives compensation from Precision Biosciences and Adaptive Biotechnologies as an Advisory Board Member. Daniel Abate-Daga receives research funding from the H. Lee Moffitt Cancer Center and Research Institute, Moffitt’s Lung Cancer Center of Excellence, and a Career Enhancement Award from NIH SPORE grant P50 CA168536.
